We are happy to announce the release of Rfam version 14.10. This release includes 62 new microRNA families and 138 updated miRNA families, 9 updated Hepatitis C Virus (HCV) families and a new intron family, the bZIP non canonical Hac1/Xbp1 intron. Read on more details.
Rfam has completed the first phase of synchronisation with miRBase
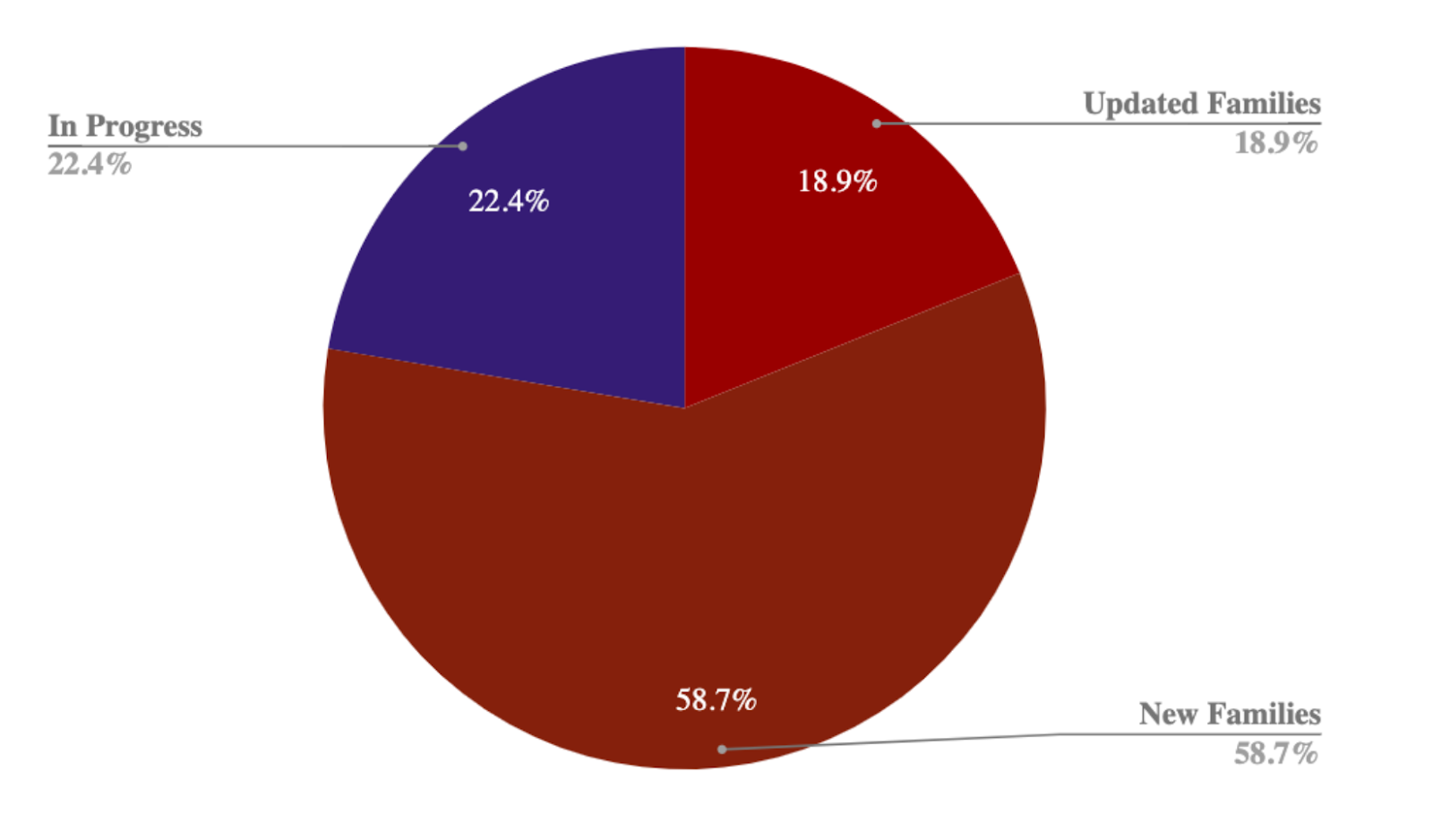
Rfam, miRBase, and RNAcentral have been working to synchronise miRNA families between all three resources. In this release we created 62 new miRBase families and updated 138. Over the course of the project we have created and updated 1536 families in total. We have updated all miRNA families in Rfam that do not require novel data from miRBase. This release completes the first phase of the synchronisation! Many thanks to everyone who has worked on this project including Joanna Argasinska, Emma Cooke, Ioanna Kalvari, Anton I. Petrov, Nancy Ontiveros-Palacios, Blake A. Sweeney and Sam Griffiths-Jones.
Further phases will focus on updating or improving miRBase families and then reflecting these changes in miRBase and Rfam. Users interested in the detailed changes to Rfam families can browse this sheet. A summary of the changes over the course of this project are:
| Release | Updated | New | Total |
| 14.3 | 37 | 353 | 390 |
| 14.4 | 0 | 496 | 496 |
| 14.5 | 112 | 0 | 112 |
| 14.6 | 0 | 126 | 126 |
| 14.7 | 121 | 0 | 121 |
| 14.8 | 30 | 25 | 55 |
| 14.9 | 22 | 14 | 36 |
| 14.10 | 138 | 62 | 200 |
| Total | 460 | 1076 | 1536 |
Hepatitis C Virus families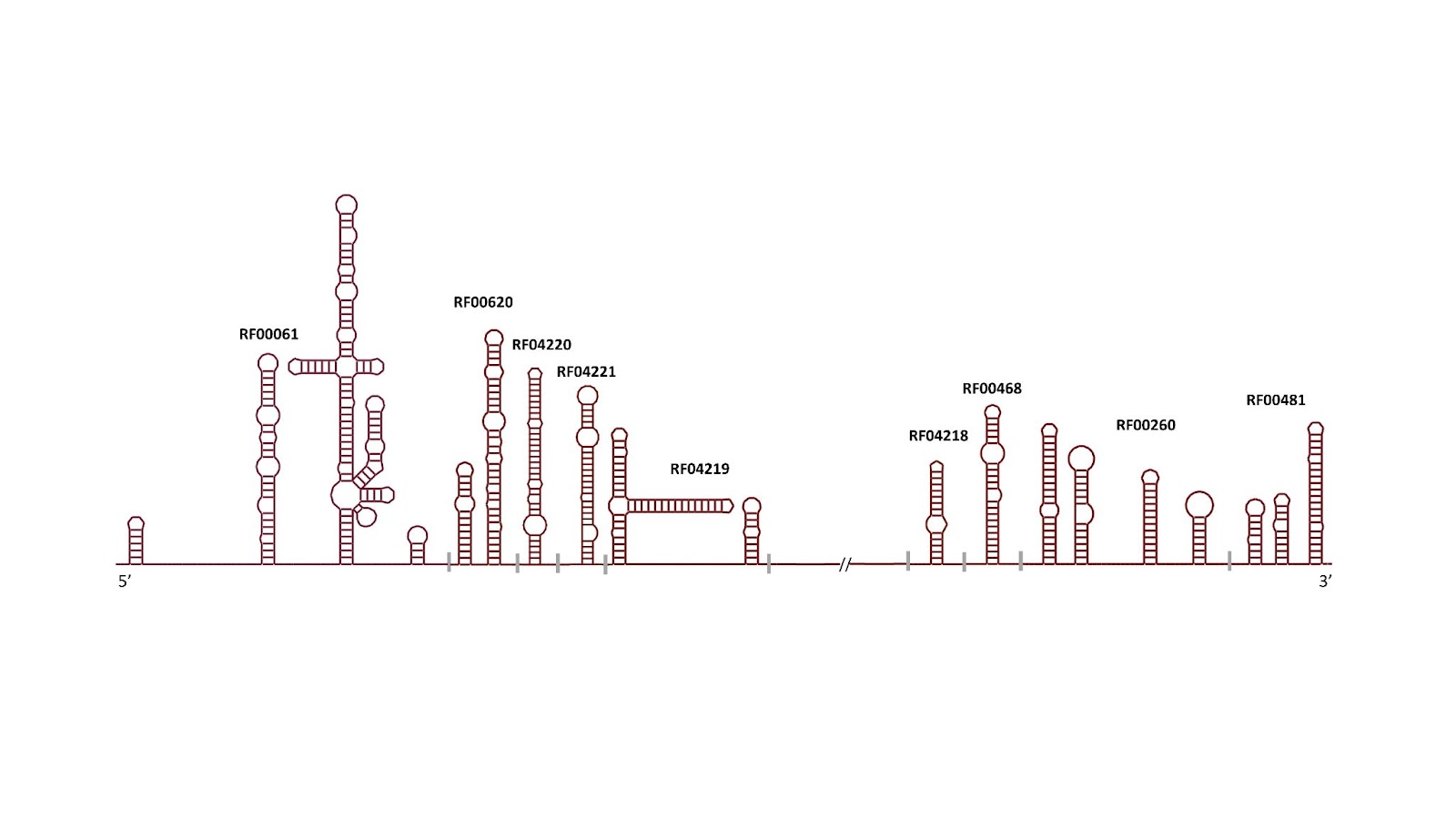
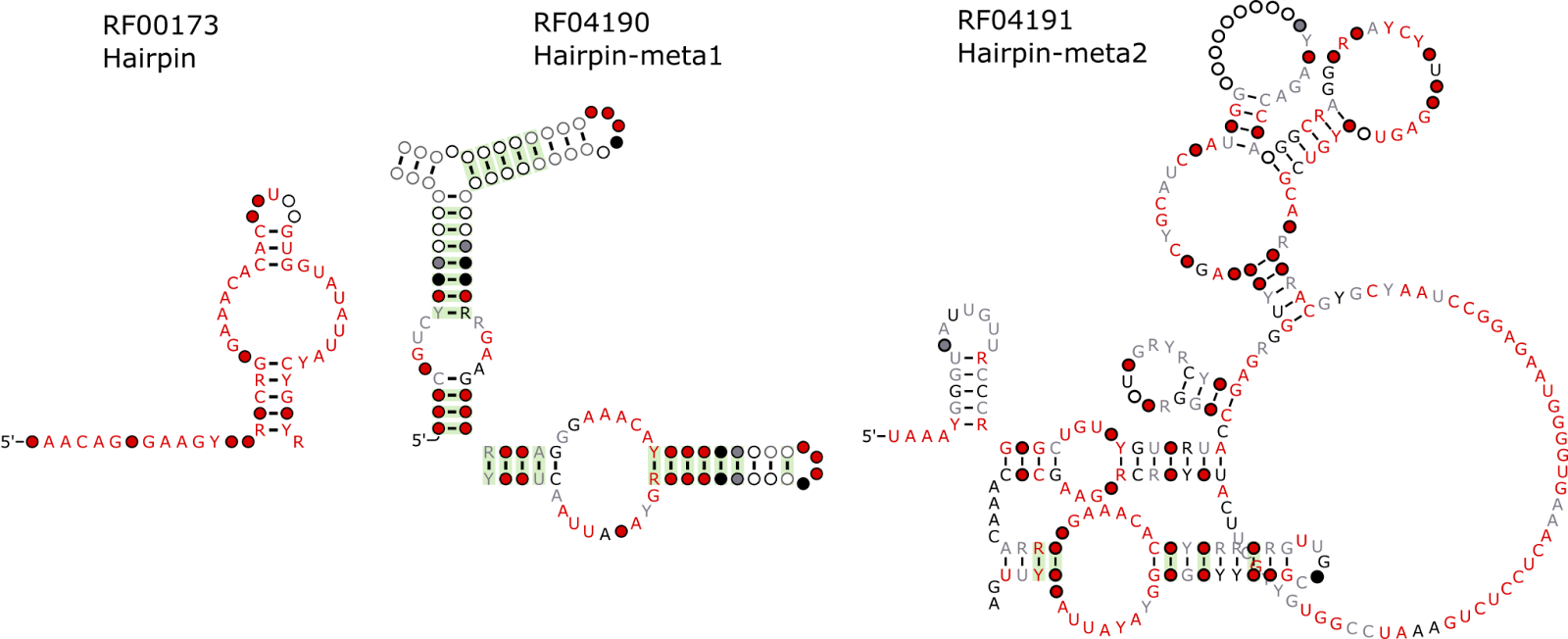
In our ongoing collaboration between Profesor Manja Marz of the European Virus Bioinformatics Center and Rfam, we have updated 9 HCV families to better reflect their secondary structures. The Marz group is planning on publishing this work shortly, keep an eye out for their paper. Thank you to Sandra Triebel and Manja Marz for their work on this. The families are listed below.
| Rfam | Family | Description |
| RF04220 | HCV_SL588 | Hepatitis C virus SL588 non-coding RNA |
| RF04221 | HCV_SL669 | Hepatitis C virus SL669 non-coding RNA |
| RF04219 | HCV_J750 | Hepatitis C virus J750 non-coding RNA (containing SLSL761 and SL783) |
| RF04218 | HCV_5BSL1 | Hepatitis C virus stem-loop I |
| RF00468 | HCV_SLVII | Hepatitis C virus stem-loop VII |
| RF00620 | HCV_ARF_SL | Hepatitis C alternative reading frame stem-loop |
| RF00260 | HepC_CRE | Hepatitis C virus cis-acting replication element (CRE) |
| RF00481 | HCV_X3 | Hepatitis C virus 3′ X element |
| RF00061 | IRES_HCV | Hepatitis C virus internal ribosome entry site |
bZIP non-canonical Hac1/Xbp1 intron
bZIP intron RNA is a non-canonical intron that was first reported in the transcriptional factor Hac1 gene in yeast and the Xbp1 gene in Metazoa. bZIP introns splice independent of the spliceosome, instead the splicing is done by ribonuclease Ire1 and guided by the secondary structure of bZIP intron. The splicing has been described in Xbp1 gene of human, mouse, Caenorhabditis elegans and Drosophila melanogaster and in Hac1 gene of fungi. The conserved structure was reported in Hooks et al, and we have created bZIP (RF04247) for this RNA.

Secondary Structure of bZIP intron. On the left, consensus structure of Hac1/Xbp1 mRNA taken from Griffiths-Jones S. & Hooks K. B. Conserved RNA structures in the non-canonical Hac1/Xbp1 intron. RNA Biology 2011; 8:4, 552-556. https://doi.org/10.4161/rna.8.4.15396. On the right RF04247 Rfam family secondary structure of bZIP intron.